Cutaneous T Cell
Lymphoma
Introduction
Lymphoma is a type of cancer that begins in a
lymphocyte. This disease is divided into two major categories: Hodgkin lymphoma
and all other lymphomas (also referred to as non-Hodgkin lymphomas).
A lymphocyte is a type of white cell. Lymphocytes
compose about 20 percent of the white cells in the blood. Most lymphocytes are
found in the lymphatic system, the major part of the body's immune system. The
lymphatic system consists of a network of organs, including the spleen, the
lymph nodes (small bean-shaped structures located throughout the body), the
lymphatic vessels and areas in the gastrointestinal tract.
The three main types of lymphocytes are: T
lymphocytes, B lymphocytes and natural killer (NK) cells. These cells circulate
throughout the body within the lymphatic vessels, suspended in a watery fluid
called lymph. The lymphatic vessels ultimately empty into the bloodstream.
Using the network of lymphatic vessels, lymphocytes are transported to
locations around the body where they are needed to
respond to infectious organisms, especially bacteria, fungi or viruses. On
contact with these infectious agents, T and B lymphocytes work in coordination
to make antibodies. The antibodies coat the infectious agents, making them
susceptible to ingestion and destruction by other white cells (called
neutrophils and monocytes). Also, NK cells can attack
virus-infected cells and help to resolve viral infections.
Most lymphomas begin in a lymphocyte in a lymph node,
when damage to cell DNA causes a cell change (mutation). The now abnormal cell
divides, and subsequent cells grow in an uncontrolled way, forming a tumor. Additional
tumors may grow in other lymph nodes or in tissues such as liver, lung and
bone. Some lymphomas begin in other lymphatic structures or in organs, such as
lymphoma of the stomach.
Cutaneous or skin lymphoma begins in a lymphocyte in
the skin. Because the medical term for the skin is the cutaneous tissue
or system, “cutaneous lymphoma” is the formal name for lymphoma of the skin.
This disease may also be called “skin lymphoma.” The term “cutaneous lymphoma”
or “skin lymphoma” actually describes a number of different disorders with
various signs and symptoms, outcomes and treatment considerations. Most skin
lymphoma begins in a T lymphocyte that has undergone a malignant change. This
disease is called “cutaneous T-cell lymphoma” or CTCL. Older descriptive terms,
such as “mycosis fungoides” and “Sézary syndrome,” are types of cutaneous
lymphomas that are now also referred to as CTCL.
Mycosis fungoides (MF) is the most common type of
CTCL. As a result, more is known
about it than other types of skin lymphomas. The name
comes from the mushroom-like skin tumors noted in the first patient diagnosed.
MF is usually a low-grade (slow-growing or indolent) lymphoma that primarily
affects the skin. It often remains confined to the skin. In general the
prognosis for MF is considered to be good. Most patients are diagnosed in early
stages with skin involvement only, and the disease does not progress to the
lymph nodes or internal organs. However, in a minority of cases MF does slowly
progress. Sézary syndrome is a related, but more aggressive form of CTCL, with
widespread skin effects and the presence of malignant lymphocytes in the blood.
This condition is characterized by an extensive red rash and sometimes loss
(sloughing) of the exterior layers of the skin. It was identified by the French
dermatologist Sézary, who first reported that some people with this skin
condition also had malignant lymphocytes in the blood.1
The overall annual incidence of primary CTCL in the US in 1988 was 0·5–1·0 per 100 000 based on population data. However, the prevalence is much higher because most patients have low-grade disease and live long. Males and the black population are affected more commonly.2,3 The incidence has increased during the past two decades but this almost certainly reflects improved diagnosis of earlier stages and possibly better registration, particularly in the US.2
Aetiology
The underlying aetiology is unknown. There is evidence for inactivation of key tumour suppressor genes and TH2 cytokine production by tumour cells in mycosis fungoides/Sezary syndrome, but no disease-specific molecular abnormality has yet been identified. Primary CTCL must be distinguished from human T-lymphotropic virus type-1 (HTLV-I) associated adult T-cell leukaemia lymphoma (ATLL) in which skin involvement often closely mimics the clinicopathological features of mycosis fungoides/Sezary syndrome and may be the presenting feature.2
Mycosis Fungoides
Mycosis fungoides represents the most common type of
cutaneous T-cell lymphoma. It is also a long-standing entity, having been
described almost two centuries ago, in 1806, by the French dermatologist
Alibert. Traditionally, it is divided into three clinical phases: patch, plaque
and tumour stages. The clinical course can be protracted over years or decades.
The term ‘mycosis fungoides’ should be restricted to the classic so-called
‘Alibert–Bazin’ type of the disease, characterized by the typical slow
evolution and protracted course. More
aggressive entities (e.g. mycosis fungoides ‘a tumeur d’emblee’), characterized
by an onset with plaques and tumours, an aggressive course and a bad prognosis,
are better classified among the recently described group of cutaneous cytotoxic
T- (NK/T-) cell lymphomas.
In the past, mycosis fungoides has been considered as
an ‘incurable’, albeit slowly progressive disease, that inevitably ended with
the death of the patient. Recently, an early
form of mycosis fungoides has been recognized,
consisting of subtle patches of the disease. These patients have relatively
mild stable disease, which questions the traditional
concept of the inevitability of disease progression
until death. The aetiology of mycosis fungoides remains unknown. A genetic
predisposition may have a role in some cases, and a familial occurrence of the
disease has been reported in a few instances. Association with long-term
exposure to various allergens has also been advocated, as well as exposure to
environmental agents and association with chronic skin disorders and viral
infections. Recently, seropositivity for cytomegalovirus (CMV) has been
observed at unusually high frequencies in patients with mycosis fungoides,
suggesting a role for this virus in the pathogenesis of the disease. In some
countries, mycosis fungoides-like disorders are clearly associated with viral
infections (human T-cell lymphotrophic virus I [HTLV-I]-associated adult T-cell
lymphoma–leukaemia), but the search for viral particles in patients with
mycosis fungoides has so far been unsuccessful. Genetic alterations have been
identified mainly in late stages of the disease, and their importance for
disease initiation is unclear.
Mycosis fungoides has been
described in patients with other haematological disorders, especially
lymphomatoid papulosis and Hodgkin lymphoma. In occasional patients, the same
clone has been detected in mycosis fungoides and associated lymphomas, raising
questions about a common origin of the diseases. In addition, patients with
mycosis fungoides are at higher risk of developing a second (non-haematological)
malignancy.
A staging classification
system for mycosis fungoides was proposed in 1979 by the Mycosis Fungoides
Cooperative Group (TNMB staging). This system takes into account the percentage
of body area covered by lesions, and the presence of lymph node or visceral
involvement. Although the presence of malignant circulating cells in the blood
should be recorded for each patient, these data are not used for staging. More
recently, a new staging classification for mycosis fungoides has been proposed
in the World Health Organization (WHO) classification of haematopoietic
neoplasms. Some centres specializing in the study and management of skin
lymphomas do not utilize the TNM or WHO staging schemes, but classify mycosis
fungoides according to the type of skin lesions (patches, plaques and tumours)
and the presence or absence of large cell transformation and/or extracutaneous involvement.
In this classify, stage I disease is confined to the skin and characterized
morphologically by patches only. Survival is extremely long in these patients
(usually decades), and nonaggressive treatments should be applied. Most
patients in this stage die of unrelated causes. Patients with stage II in this classify
also have disease limited to the skin, but characterized morphologically by the
presence of plaques, tumours or erythroderma, or by large cell transformation
histopathologically. The disease in these patients is inevitably progressive, and
treatment should be more aggressive. Stage III patients have extracutaneous
disease and should be managed with aggressive treatment options. Staging
investigations are not necessary in early stage mycosis fungoides (patch
stage). Patients with plaques, tumours or erythroderma should be screened for
extracutaneous involvement (laboratory investigations, sonography of
lymphnodes, computerized tomography (CT) scan of thorax and abdomen, bone
marrow biopsy, examination of the peripheral blood). Although the presence of a
monoclonal population of T lymphocytes within the peripheral blood has been
observed by polymerase chain reaction (PCR) technique in some patients with early mycosis fungoides, in
many of these cases the clone was different
from that detected in the skin lesions. In addition, the prognostic value of
the detection of monoclonality in the peripheral blood is unclear. It has been
suggested that flow cytometry analysis is highly effective in demonstrating and
quantifying small numbers of circulating tumour cells in patients with mycosis
fungoides.4
Lesions of mycosis fungoides can be divided
morphologically into patches, plaques and tumours. Itching is often a prominent
symptom. Erythroderma may develop in the course of the disease, rendering
distinction from Sézary syndrome difficult without a proper clinical history.4,5,6,7
Patch stage
Patches of mycosis fungoides are characterized by
variably large, erythematous, finely scaling lesions with a predilection for
the buttocks and other sun-protected areas . Loss of elastic fibres and atrophy
of the epidermis may confer on the lesions a typical wrinkled appearance, and
terms such as ‘parchment-like’ or ‘cigarette paper-like’ have been used to describe them. Sometimes, these single patches
have a yellowish hue, conferring a ‘xanthomatous’-like aspect to the lesions
(xanthoerythroderma perstans). In early phases, a ‘digitate’ pattern can be
observed (alone or in combination with larger patches; see also smallplaque
parapsoriasis).4,7,8
Plaque stage
Plaques of mycosis fungoides are characterized by
infiltrated, scaling, reddish brown lesions. Typical patches are usually
observed contiguous to plaques or at other sites on the body. Plaques of
mycosis fungoides should be distinguished from flat tumours of the disease. Flat
infiltrated lesions should be biopsied in order to allow histopathological
examination and a precise classification of the lesions.4,7,8
Tumour stage
In tumour-stage mycosis fungoides a combination of
patches, plaques and tumours is usually found, but tumours may also be observed
in the absence of other lesions. Tumours may be solitary or, more often,
localized or generalized. Ulceration is common. In tumour-stage mycosis
fungoides unusual sites of involvement may be observed, such as the mucosal
regions. As oral and genital mucosae are frequently involved in cytotoxic
T/NK-cell lymphomas, care should be taken to classify these cases correctly.
Careful clinical history taking, re-evaluation of previous biopsies, and
complete phenotypical and genotypical investigations are mandatory to make the
diagnosis of mucosal involvement in mycosis fungoides.4,7,8
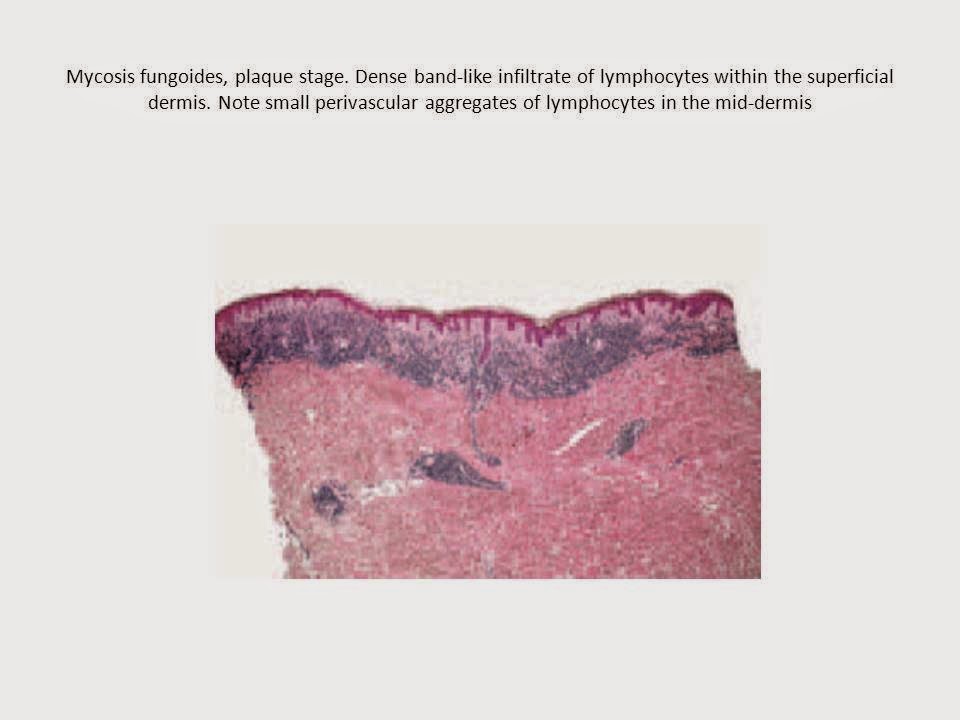
Histopathology
Early lesions of mycosis fungoides reveal a patchy
lichenoid or band-like infiltrate in an expanded papillary dermis. A
psoriasiform hyperplasia of the epidermis may be seen , but in most cases the
epidermis is normal. Small lymphocytes predominate, and atypical cells
can be observed only in a minority of cases.
Epidermotropism of solitary lymphocytes is usually found, but Darier’s nests
(Pautrier’s microabscesses) are rare. Useful diagnostic clues are the presence
of epidermotropic lymphocytes with nuclei slightly larger than those of
lymphocytes within the upper dermis and/or the presence of lymphocytes aligned along the basal layer of the
epidermis. Also useful is the presence of many intraepidermal lymphocytes in
areas with scant spongiosis. In this context, it should be emphasized that in a
few cases (approximately 5% of the total) epidermotropism may be missing. The
papillary dermis shows a moderate to marked fibrosis with coarse bundles of
collagen and a band-like or patchy lichenoid infiltrate of lymphocytes. Dermal
oedema is usually not found. Unusual histopathological patterns of mycosis
fungoides in early phases include the presence of a perivascular (as opposed to
band-like) superficial infiltrate, prominent spongiosis simulating the picture
of acute contact dermatitis, an interface dermatitis, sometimes with several necrotic
keratinocytes, marked pigment incontinence with melanophages in the papillary
dermis, prominent epidermal hyperplasia simulating the picture of lichen
simplex chronicus and prominent extravasation of erythrocytes. A pattern characterized
by a markedly flattened epidermis, a lichenoid infiltrate in the dermis and
increased dilated vessels in the papillary dermis is the histopathological
counterpart of poikilodermatous mycosis fungoides. Cytomorphologically, small
pleomorphic (cerebriform) cells predominate. In some cases, plaques or flat
tumours of mycosis fungoides may present with a predominantly interstitial
infiltrate. This peculiar presentation can give rise to diagnostic problems,
and has been designated ‘interstitial mycosis fungoides’. Immunohistology
confirms that interstitial cells are T lymphocytes, thus being a helpful clue
for the differential diagnosis with the interstitial variant of granuloma
annulare. Intersitial mycosis fungoides is usually a manifestation of either
the plaque or tumour stage of the disease. In tumours of mycosis fungoides, a
dense nodular or diffuse infiltrate is found within the entire dermis, usually
involving the subcutaneous fat. Epidermotropism may be lost. Flat tumours are
characterized histopathologically by dense infiltrates confined to the
superficial and mid parts of the dermis. Angiocentricity and/or
angiodestruction can be observed in some cases. A peculiar histopathological
presentation of mycosis fungoides characterized by marked involvement of
hyperplastic sweat glands has been termed ‘syringotropic’ mycosis fungoides. In
some of these cases, syringometaplasia can be observed. Involvement of the
epidermis may be missing in syringotropic mycosis fungoides, thus creating
problems in the histopathological diagnosis of this variant of the disease.4,6,7
Immunophenotype
Mycosis fungoides is characterized by an infiltrate
of α/β T helper memory lymphocytes (βF1+, CD3+, CD4+, CD5+, CD8–, CD45RO+).
Only a minority of cases exhibit a T-cytotoxic (βF1+, CD3+, CD4–, CD5+, CD8+)
or γ/δ (βF1–, CD3+, CD4–, CD5+, CD8+) lineage that show no clinical and/or
prognostic differences. In these cases, correlation with the clinical features
is crucial, in order to rule out skin involvement by aggressive cytotoxic
lymphomas such as CD8+ epidermotropic T-cell lymphoma or γ/δ T-cell lymphoma.
In late stages there may be a (partial) loss of pan-T-cell antigen expression.
In
plaque and tumour lesions, neoplastic T cells may
express the CD30 antigen. Recently, it has been suggested that a low CD8 : CD3
ratio in skin infiltrates supports the histopathological diagnosis of mycosis
fungoides, but this finding should be confirmed by larger studies.
Cytotoxic-associated markers such as TIA-1 and granzyme B are negative in
mycosis fungoides, although occasionally in late stages of the disease some
positivity may be observed. These cases should not be classified as cytotoxic
lymphomas, but as tumour-stage mycosis fungoides with cytotoxic phenotypes. A
similar phenotype may also be seen in early lesions of the rare γ/δ+ mycosis
fungoides, which besides cytotoxic proteins also express CD56.4
Histopathological Differential Diagnosis
The histopathological diagnosis of early mycosis
fungoides may be extremely difficult. In some instances, differentiation from
inflammatory skin conditions (e.g. psoriasis, chronic contact dermatitis) may
be impossible on histopathological grounds alone. In these cases, clinical
correlation is crucial to make a definitive diagnosis. Immunohistological
features are not distinctive, and are similar to those observed in many
inflammatory skin conditions. Staining for CD3 or CD4 may help by highlighting
epidermotropic T lymphocytes.4
Sézary syndrome
Sézary syndrome is characterized clinically by
pruritic erythroderma, generalized lymphadenopathy and the presence of
circulating malignant T lymphocytes (Sézary cells).4,5 Other typical
cutaneous changes include palmoplantar hyperkeratosis, alopecia and
onychodystrophy. Differentiation from non-neoplastic erythroderma may be
extremely difficult. The main causes of erythroderma, besides cutaneous T-cell
lymphoma, are atopic dermatitis, psoriasis and drug reactions. Erythrodermic
mycosis fungoides should be distinguished from true Sézary syndrome.
The presence of a monoclonal population of T
lymphocytes within the peripheral blood has been recently proposed by the
European Organization for Research and Treatment of Cancer (EORTC) Cutaneous
Lymphoma Study Group, by the International Society for Cutaneous Lymphomas and
by others as an important criterion for the diagnosis of Sézary syndrome. Other
useful criteria include the presence of more than 1000 circulating Sézary cells/mm;
an expanded CD4+ population in the peripheral blood resulting in a markedly
increased CD4+ : CD8+ ratio (> 10); an increased population of CD4+ : CD7–
cells in the peripheral blood; Sézary cells larger than 14 μm in diameter; and
Sézary cells representing more than 20% of circulating lymphocytes.
Patients usually present with an abrupt onset of
erythroderma, or with erythroderma preceded by itching and a nonspecific skin
rash. Rarely, a classic Sézary syndrome may develop in patients with preceding
mycosis fungoides; it has been suggested to classify these cases as ‘Sézary
syndrome preceded by mycosis fungoides’, as it remains unclear whether the
clinical features and prognosis are similar. The presence of neoplastic T cells
within the peripheral blood alone should not prompt a diagnosis of Sézary
syndrome unless all other main diagnostic criteria are met.
The TNM staging classification used for mycosis
fungoides has also been adopted for Sézary syndrome. According to this system,
Sézary syndrome is classified as stage III by definition.4
The histopathological features of skin lesions in
Sézary syndrome are indistinguishable from those of mycosis fungoides. Often
there is a psoriasiform spongiotic pattern with a variably dense band-like
infiltrate of lymphocytes. Epidermotropism is usually less marked than in mycosis
fungoides, but typical Darier’s nests ‘Pautrier’s collections’ may be observed.
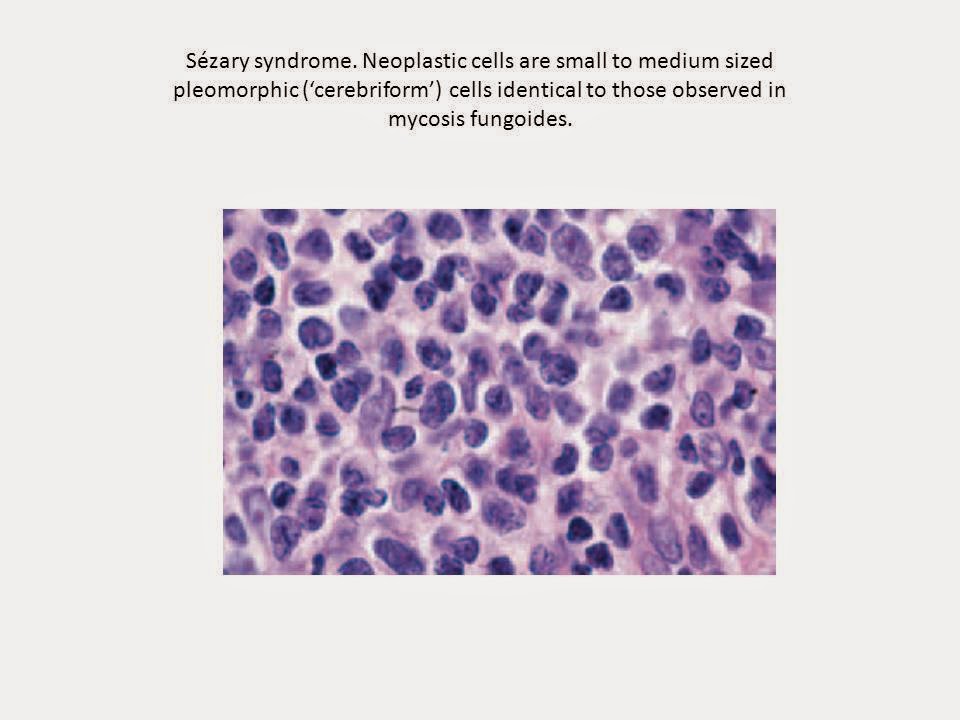
Cytomorphology reveals a predominance of small to medium-sized pleomorphic
(cerebriform) lymphocytes, often referred to as ‘Sézary cells’. Differential
diagnosis from mycosis fungoides can be achieved only by correlation of
histopathological features with clinical ones. Histopathological variants of
Sézary syndrome include the presence of a prominent granulomatous reaction,
deposition of mucin within hair follicles (follicular mucinosis), or large cell
transformation. Large cell transformation may be detected in skin lesions,
lymph nodes, or both, and is indistinguishable from that occurring in advanced
mycosis fungoides. The lymph nodes are characterized by monomorphous
infiltrates of neoplastic cells, and reveal histopathological differences from
lymph nodes involved by cells of mycosis fungoides, suggesting a pathogenetic
difference between the two disease.4
Prognosis
Most cases of primary CTCL are not curable. Independent prognostic features in mycosis
fungoides include the cutaneous and lymph node stage of disease and age of onset (>60
years). The lymph node status and tumour burden within peripheral blood determine the
prognosis in Sezary syndrome. Serum lactate dehydrogenase and the thickness of the infiltrate in plaque-stage mycosis fungoides are also independent markers of prognosis. Multivariate analysis indicates that an initial complete response (CR) to various therapies is an independent favourable prognostic feature, particularly in early stages of disease. For mycosis fungoides, two staging systems are in regular use including a TNM (primary tumour, regional nodes, metastasis) system and a clinical staging specifically designed for CTCL. These staging systems can also be applied to Sezary syndrome, but neither system provides a quantitative method for assessing peripheral blood disease other than an additional B0 and B1 in the TNM system and this has prompted alternative approaches for Sezary syndrome. The 5- and 10-year overall survival (OS) in mycosis fungoides are 80% and 57%, respectively, with disease-specific survival (DSS) rates of 89% and 75% at 5 and 10 years respectively. Patients with very early stage disease (IA) are highly unlikely to die of their disease, with DSS rates of 100% and 97–98% at 5 and 10 years, respectively and risks of disease progression varying from 0% to 10% over 5–20 years.2,9
Treatment
Treatment for CTCL depends on the type and stage of
the disease. Treatment options include phototherapy, radiation, topical
therapy, systemic single-agent chemotherapy, combination chemotherapy and
combined therapies. Patients with localized early-stage disease are usually
treated with topical agents such as nitrogen mustard, skin-softening agents,
anti-itch agents and gradual exposure to sunlight or ultraviolet light.
Specific treatments include:
- Topical
nitrogen mustard
Nitrogen mustard or
Mustargen® is a chemotherapeutic agent that is administered as an ointment, an
aqueous solution or a liquid gel formulation. It is applied daily either to
affected areas of skin or to all skin surfaces. People in the early stage of
the disease respond best, but relapses are common after therapy is stopped.
- Photochemotherapy
Psoralen is a drug that
binds to the DNA in malignant cells. Patients take the drug by mouth and then
are exposed to ultraviolet light to activate the drug, which damages the
malignant cell DNA. This treatment often is referred to as PUVA, an acronym for
Psoralen and Ultraviolet-A light. Treatment is usually given several times a
week for one or two months, and less frequently thereafter. Maintenance treatment
is usually continued for a year or more. Patients in the early phase of disease
have the best response to treatment. Low-dose alpha-interferon, combined with
PUVA therapy, has shown promising preliminary results for patients with
early-stage disease. Narrow-band UVB, a form of phototherapy, is also used to
treat patch-stage cutaneous lymphoma.1,9,10
- Electron
beam radiation
Conventional radiation
therapy penetrates the skin and reaches areas inside the body. Electron beam
therapy can be applied to the entire skin surface without affecting internal
organs. This type of radiation has been helpful for patients who have skin
tumors. The tumors often heal after treatment and the dead tissue resolves,
reducing the risk of infection. Treatment leads to complete clearing of lesions
in several months without further treatment. Some patients appear to have
longstanding regression of their skin lesions following this type of treatment.
- Chemotherapy
Several combinations of
chemotherapeutic agents have been used in patients with skin lymphoma,
especially those with disease that involves the lymph nodes or other organs, or
those with Sézary syndrome. Effective therapies include methotrexate,
gemcitabine and liposomal anthracyclines. Other agents such as etoposide
(VP-16), cyclophosphamide and pentostatin may also be used. The most commonly
used drug combinations include cyclophosphamide, doxorubicin, vincristine and
prednisone.This particular drug combination may also be used for those patients
experiencing disease transformation or organ involvement. Chemotherapy has not
been able to cure widespread skin lymphoma, and studies using chemotherapy
combined with radiation therapy in patients with early stages of disease have
not been very successful.
- Other
drug therapies
Skin lymphomas are covered
under the U.S. Orphan Drug Act (1983). The Orphan Drug Act provides incentives
to induce sponsors to develop and manufacture drugs that otherwise might not be
profitable because of their small potential market. Since 1983, more than 200
drugs and biological products for rare diseases have been brought to market,
compared with 10 such products in the decade prior to 1983.
- Photopheresis
This procedure, also
called ECP for “extracorporeal phototherapy,” is a form of chemotherapy that
requires removal of blood through a vein and isolation of the white cells,
which includes circulating CTCL cells. The removed cells are treated with a
drug, 8-methoxsalen (UVADEX®), which sensitizes the cells to ultraviolet light.
An external source of ultraviolet A (UVA) rays is used to irradiate the cells,
which are then returned to the patient
through a vein. The ultraviolet light acting on 8-methoxsalen damages DNA of
the CTCL cells. The procedure must be repeated multiple times to gain the full
effect. It is believed that the effect is more extensive than killing removed
CTCL cells. It has been postulated that it involves a reaction of normal immune
cells against the UV light-damaged lymphoma cells. Thus, there may also be an
immune component because more CTCL cells are destroyed than the number
irradiated in the procedures. Photopheresis is most often used for advanced
stages of CTCL with signs of circulating CTCL cells in the blood. It is often
combined with other therapies. Photopheresis is also being studied in clinical
trials for early-stage disease.
- Supportive
therapy
The itching that accompanies
the skin lesions can be difficult to control. Antihistamines, particularly
Benadryl® or Atarax®, may relieve itching to someextent. However, the major
side effect of these drugs is drowsiness. Also, patients may develop a
resistance (tolerance) to the effectiveness of the drugs and require larger
doses. Application of skin softeners or steroid ointments may also help relieve
itching. Antibiotics are given if lesions become infected. Patients with
long-standing, troublesome symptoms may require treatment for depression or
insomnia.1,7,9,11,12
References
1. cutaneus T Cell Lymphoma. Available at: www.leukemia-lymphoma.org/attachments/.../br_1163608564.pdf.
2.
Williams H. Bigby M. Diepgen T. Herxheimer A. Naldi L.
Rzany B. Evidence Based Dermatology: Primary Cutaneus T cell Lymphoma. BMJ.
London. 2003 : 344-345.
3.
Twenty-Year Trends in the Reported Incidence
of Mycosis Fungoides and Associated Mortality. Available at: www.ajph.org/cgi/content/abstract/89/8/1240.
4.
Cerroni
L. Gatter K. Kerl H. An Illustrated Guide to Skin Lymphoma: Cutaneus T Cell
Lymphoma. Blackwell Publishing Ltd. USA
5.
Kumar V, Abbas A.K, Fausto N. Pathologic Basis of
Disease: Mycosis Fungoides. Elsevier Saunders. Philadelphia . 2005: 685.
6.
Raphael R. Strayer D.S. Rubin’s Pathology:
Clinicopathologic Foundations of Medicine. Lippincott Williams & Wilkins.
Philadephia. 2008: 921.
7.
Pillsbury D.M. Shelley W.B. Kligman A.M. Dermatology:
The Lymphoma. W.B.Saunders Company. Philadelphia .
1960: 1089-1093.
8.
Girardi M. Heald P.W. Wilson L.D. The Pathogenesis of
Mycosis Fungoides. NEJM. Volume 350: 1978-1988.
9.
Suarez Varela M.M.M. Cutaneus T Cell Lymphoma. CRC
Press. 2005: 1-32.
10.
The Addition of Interferon
Gamma to Oral Bexarotene Therapy With
Photopheresis for Sézary Syndrome. Available at: http://archderm.ama-assn.org/cgi/content/extract/141/9/1176.
11.
Bickers D. Henry W.Lim.
Margolis D. Weinstock M. The Burden of Skin Disease. The Lewin Group Ltd. Washington . 2004: 29-31.
12.
Classification
and Treatment of Rare and Aggressive Types of
Peripheral T-Cell/Natural Killer-Cell
Lymphomas of the Skin. Available at: www.moffitt.org/moffittapps/ccj/v14n2/pdf/112.pdf.

















I'm 55-year-old from Paris, I was diagnosed with second-stage liver cancer following a scheduled examination to monitor liver cirrhosis. I had lost a lot of weight. A CT scan revealed three tumors; one in the center of my liver in damaged tissue and two in healthy portions of my liver. No chemotherapy or radiotherapy treatment was prescribed due to my age, the number of liver tumors. One month following my diagnosis I began taking 12 (350 point) Salvestrol supplements per day, commensurate with my body weight. This comprised six Salvestrol Shield (350 point) capsules and six Salvestrol Gold (350 point) capsules, spread through the day by taking two of each capsule after each main meal. This level of Salvestrol supplementation (4,000 points per day) was maintained for four months. In addition, I began a program of breathing exercises, chi exercises, meditation, stretching and stress avoidance. Due to the variety of conditions that I suffered from, I received ongoing medical examinations. Eleven months after commencing Salvestrol supplementation But all invalid so I keep searching for a herbal cure online that how I came across a testimony appreciating Dr Itua on how he cured her HIV/Herpes, I contacted him through email he listed above, Dr Itua sent me his herbal medicine for cancer to drink for two weeks to cure I paid him for the delivering then I received my herbal medicine and drank it for two weeks and I was cured until now I'm all clear of cancer, I will advise you to contact Dr Itua Herbal Center On Email...drituaherbalcenter@gmail.com. WhatsApps Number...+2348149277967. If you are suffering from Diseases listed below,
BalasHapusCancer
HIV/Aids
Herpes Virus
Bladder cancer
Brain cancer
Colon-Rectal Cancer
Breast Cancer
Prostate Cancer
Esophageal cancer
Gallbladder cancer
Gestational trophoblastic disease
Head and neck cancer
Hodgkin lymphoma
Intestinal cancer
Kidney cancer
Leukemia
Liver cancer
Lung cancer
Melanoma
Mesothelioma
Multiple myeloma
Neuroendocrine tumors
Non-Hodgkin lymphoma
Oral cancer
Ovarian cancer
Sinus cancer
Skin cancer
Soft tissue sarcoma
Spinal cancer
Stomach cancer
Testicular cancer
Throat cancer
Thyroid Cancer
Uterine cancer
Vaginal cancer
Vulvar cancer
Hepatitis
Chronic Illness
Lupus
Diabetes
Fibromyalgia.